6 دقیقه
سلولها برای تأمین انرژی به میتوکندریها وابستهاند، اما پژوهشهای جدید نشان میدهد خطاهای مرتبط با تکثیر DNA میتوکندریایی (mtDNA) میتواند به التهاب مزمنِ مرتبط با پیری منجر شود. محققان مؤسسه ماکس پلانک برای زیستشناسی پیری و همکارانشان با بررسی نمونههای بافتی انسانی و مدلهای موشیِ مرتبط با پیری، مکانیزمی مولکولی را شناسایی کردند که باعث میشود میتوکندریها بخشهایی از DNA خود را بیرون ریخته و این قطعات خارجشده، پاسخهای التهابی را در سلولها فعال کنند.
پیشزمینه علمی: mtDNA، نوکلئوتیدها و پیری سلولی
میتوکندریها دارای ژنوم مجزایی به نام mtDNA هستند که باید در طول فرایند نگهداری و بازتکثیر بهدرستی نسخهبرداری شود. در سلولهای هستهای، واحدهای سازندهٔ تکثیر DNA نوکلئوتیدهای دئوکسیریبونوکلئوتیدی (dNTPها) هستند؛ موجودی و تعادل این dNTPها برای حفظ صحت نسخهبرداری حیاتی است. مطالعات پیشین نشان دادهاند که ذخیرهٔ dNTPها در سلولهای پیر یا سنسِنت کاهش مییابد و این میتواند کیفیت سنتز DNA را به مخاطره بیندازد.
هرگاه دئوکسیریبونوکلئوتیدها کم شوند، پلیمرازهای میتوکندریایی ممکن است بهاشتباه از ریبونوکلئوتیدها (rNTPها) استفاده کنند. وارد شدن ریبونوکلئوتیدها به ساختار DNA پایداری رشته را تضعیف میکند و میتواند منجر به نقایص، شکستهای موضعی یا رونویسی ناقص شود. میتوکندری ممکن است این نسخههای ناپایدار را بهعنوان مولکولهای معیوب شناسایی کرده و آنها را به سیتوزول خارج کند. mtDNA در سیتوزول توسط حسگرهای ایمنی ذاتی شناسایی میشود و این شناسایی میتواند مسیرهای التهابی را فعال کند که در بروز آسیب بافتی و فیبروز نقش دارند.
این فرایند نه تنها یک نقص مکانیکی در نسخهبرداری را نشان میدهد، بلکه پیوندی میان وضعیت متابولیک سلول—بهویژه تعادل نوکلئوتیدها—و پاسخ ایمنی مزمن برقرار میکند. همین ارتباط میتواند توضیح دهد که چرا در طول دههها التهاب سطح پایین و مزمن در بافتها تجمع مییابد و چگونه این التهاب به بیماریهای مرتبط با سن کمک میکند.
طراحی مطالعه و یافتههای کلیدی
محققان با ترکیب نمونههای بافتی انسانی و مدلهای موشی مهندسیشده که تغییرات مرتبط با پیری را شبیهسازی میکردند، مسیر تأثیر اختلال تعادل نوکلئوتیدی بر سلامت mtDNA و التهاب سلولی را دنبال کردند. آنها از مجموعهای از روشها شامل آزمایشهای بیوشیمیایی برای اندازهگیری سطوح نوکلئوتیدها، تعیین توالی (sequencing) برای شناسایی جایگذاریهای ریبونوکلئوتیدی در mtDNA و تحلیلهای بافتشناسی برای مشاهدهٔ تغییرات ساختاری و التهابی در بافتها استفاده کردند.
نتایج نشان داد که در بافتهای پیر یا آنهایی که از نظر نوکلئوتیدی تخلیه شده بودند، نرخ وارد شدن rNTP به mtDNA بهطور قابلتوجهی افزایش یافته است. چنین mtDNAهای ناقص و حاوی ریبونوکلئوتید بیشتر در معرض جداسازی از غشای میتوکندری و خروج به سیتوزول قرار داشتند. در سطح مولکولی، این خروج mtDNA بهعنوان محرکی برای فعالشدن مسیرهای ایمنی ذاتی عمل میکرد و مجموعهای از مولکولهای التهابی و سیگنالدهی التهابی را القا نمود.
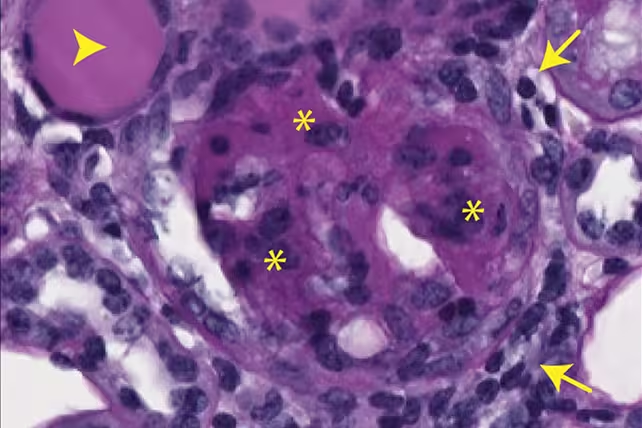
شواهد فیبروز کلیه در موشها که ناشی از mtDNA خارجشده است. (Max Planck Institute for Biology of Ageing)
در مدلهای حیوانی مشاهده شد که این فعالشدن ایمنی منجر به التهاب مزمن با ویژگیهایی مثل تشکیل بافت جوشگاهی (fibrosis) و آسیب عملکردی در بافتهایی مانند کلیهها شد. این یافتهها پیوند روشنی بین تغییرات متابولیک—بهویژه کاهش دسترسی به dNTPها—و زنجیرهای از رویدادهای مولکولی که از ناپایداری میتوکندریایی تا فنوتیپهای التهابی در بافتهای پیر را توضیح میدهد، فراهم میکنند.
علاوه بر این، تیم تحقیقاتی با استفاده از تعیین توالی عمیق و آنالیز جهشها توانست امضای مشخصی از جایگذاریهای ریبونوکلئوتیدی ارائه دهد که نشان میداد این فرایند بهویژه در مناطقی از mtDNA که مسئول نگهداری و تنظیم عملکردهای کلیدی میتوکندری هستند، متمرکز است. در نتیجه، نهتنها مقدار mtDNA بلکه کیفیت و محل خطاهای واردشده تعیینکننده شدت پاسخ التهابی بودند.
بیانات برجسته از سوی محققان
توماس لانگر از مؤسسه ماکس پلانک برای زیستشناسی پیری خلاصه میکند که اختلالات متابولیک که مخازن نوکلئوتیدی را تغییر میدهند میتوانند سلسلهای از رویدادها را آغاز کنند که در نهایت به التهاب در سلولهای سنسِنت و اندامهای پیر میانجامد؛ این نکته مسیرهای مولکولی جدیدی را بهعنوان اهداف بالقوهٔ مداخله نشان میدهد. دوسانکا میلینکوویچ، نیز از همان مؤسسه، به این اشاره میکند که درمانهایی که هماکنون برای برخی اختلالات میتوکندریایی استفاده میشوند—از جمله تأمین پیشمادههای DNA—میتوانند بررسی شوند تا مشخص شود آیا قادر به کاهش التهاب مرتبط با پیری نیز خواهند بود یا خیر.
محققان تأکید دارند که چنین راهکارهایی باید با دقت آزمایش شوند، زیرا تنظیم مصنوعی سطح نوکلئوتیدها میتواند اثرات جانبی بیوشیمیایی و ژنومی داشته باشد. به همین دلیل مطالعات پیشبالینی کنترلشده و سپس آزمایشهای بالینی ضروری هستند تا کارآیی و ایمنی چنین رویکردهایی بهدرستی سنجیده شود.
پیامدها برای سلامت و مسیرهای پژوهشی آینده
این مکانیزم، توضیحی منطقی برای چگونگی تجمع التهاب سطح پایین و مزمن در طول دههها ارائه میدهد؛ التهابی که در بسیاری از بیماریهای مرتبط با سن نقش دارد، از جمله بعضی سرطانها و اختلالات نورودژنراتیو مانند بیماری آلزایمر. اگر پژوهشگران بتوانند از وارد شدن ریبونوکلئوتیدها به mtDNA جلوگیری کنند یا ثبات mtDNA را بهگونهای افزایش دهند که از خروج آن به سیتوزول جلوگیری شود، ممکن است بتوانند سیگنالدهی التهابی مضر در بافتهای پیر را کاهش دهند.
از منظر کاربردی، چند مسیر تحقیقاتی مهم روشن میشود که میتواند به عنوان قدمهای بعدی دنبال شود:
1) تعیین فراوانی این مسیر در پیر شدن طبیعی انسان در مقایسه با وضعیتهای بیماریزای خاص. لازم است مشخص شود آیا این پدیده در همهٔ بافتها رخ میدهد یا در برخی بافتها (مانند کلیه، مغز یا بافتهای ماهیچهای) بارزتر است.
2) بررسی امکان بازگرداندن یا تثبیت مخازن dNTP در سلولهای بالغ بهصورت ایمن. این شامل آزمونهای دارویی یا تغذیهای برای افزایش پیشسازههای DNA و همچنین پیبردن به اثرات سیستمیک آنها خواهد بود.
3) کاوش در مسیرهای حسگر ایمنی که mtDNA سیتوزولی را شناسایی میکنند (برای مثال مسیرهای cGAS–STING و دیگر سامانههای تشخیص DNA) تا ببینیم آیا میتوان با مهار هدفمند این مسیرها پاسخ التهابی را تعدیل کرد بدون اینکه ایمنی ذاتی بهطور کلی تضعیف شود.
4) تعیین پیامدهای طولانیمدت مداخلات احتمالی: حتی اگر بتوان ورود rNTPها را کاهش داد یا خروج mtDNA را مهار کرد، باید اثرات جانبی ژنومی و متابولیک این مداخلات در انسان بالغ و پیر بهدقت مطالعه شود.
بهطور کلی، تمرکز بر حفظ یکپارچگی ژنوم میتوکندریایی بهعنوان استراتژیای برای کاهش التهاب مزمن میتواند یک رویکرد نوآورانه برای مقابله با مجموعهای از بیماریهای مرتبط با سن ارائه دهد. با این حال، چالش اصلی این است که این راهکارها باید طوری تنظیم شوند که عملکرد طبیعی میتوکندری و نیازهای متابولیک سلول را مختل نکنند.
جمعبندی
این مطالعه ارتباطی واضح بین کمبود متابولیک—کاهش دئوکسیریبونوکلئوتیدها—و مسیر مولکولی ایجاد التهاب از طریق ناپایداری و خروج mtDNA ارائه میدهد. این یافتهها راههایی را برای استراتژیهای هدفمند بهمنظور حفظ یکپارچگی ژنومی میتوکندریایی و کاهش افت ناشی از التهاب در جمعیتهای پیر باز میکنند. با این همه، برای تعیین اینکه آیا تقویت مخازن نوکلئوتیدی یا دیگر مداخلات میتوانند بهصورت عملی و ایمن به سود سلامت انسان منجر شوند، پژوهشهای پیشبالینی و کارآزماییهای بالینی بیشتری لازم است.
در نهایت، این پژوهش نه تنها یک پل بین فرایندهای متابولیک و ایمنی ذاتی برقرار میکند، بلکه نشان میدهد که چگونه تغییرات تدريجی در سوخت و ساز سلولی طی دههها میتواند پیامدهای گستردهتری برای سلامت ارگانیسم داشته باشد. درک دقیقتر این زنجیرهٔ علت و معلولی میتواند به توسعهٔ درمانهایی منجر شود که نه تنها بهصورت علامتی التهاب را کاهش دهند، بلکه ریشههای مولکولی آن را هدف قرار دهند.





.webp "نخستین نشانه جدی از قمر فراخورشیدی عظیم دوردست")






نظر بگذارید
نظرات
هنوز نظری ثبت نشده. اولین نفر باشید.