
7 دقیقه
چیزی بسیار ریز، دستدادن مرگباری را درون سلولهای مغزی مختل کرد. نتیجه این اختلال: میتوکندریها — نیروگاههای سلولی — شروع به نارسایی کردند و نورونهایی که پیشتر با انرژی کار میکردند، توان خود را از دست دادند. این جفت رخداد در مرکز یک مطالعه جدید از دانشکده پزشکی دانشگاه Case Western Reserve قرار دارد که یک مسیر مستقیم از تودههای آلفا-سینوکلئین تا فروپاشی میتوکندری را ردیابی میکند و طعمه مولکولیای ارائه میدهد که به توقف این آسیب کمک میکند.
آلفا-سینوکلئین برای چند دهه بهعنوان یکی از مظنونان اصلی در بیماری پارکینسون مطرح بوده است. زمانی که این پروتئین دچار تاخوردگی نابهنجار و تجمع میشود، نورونها دچار اختلال میگردند. جدا از این، پزشکان و محققان آزمایشگاهی مشاهده کردهاند که میتوکندریهای نورونهای آسیبدیده ضعیف، کمانرژی و مستعد القای مرگ سلولی هستند. اما چگونگی ارتباط این دو پدیده تا کنون مبهم بود. کار جدید یک تعامل بیوشیمیایی دقیق را روشن میکند: آلفا-سینوکلئین به آنزیمی عمیق در میتوکندری به نام ClpP میچسبد و به نظر میرسد این پیوند توانایی ارگانل در مدیریت پروتئینهای آسیبدیده و حفظ تعادل متابولیک را مختل میکند؛ یعنی همان فرایندهایی که برای سلامت انرژی نورون حیاتیاند.
یافتهها و روشهای آزمایشی تیم تحقیق
در بافتهای آزمایشگاهی و مدلهای حیوانی، پژوهشگران مشاهده کردند که آلفا-سینوکلئین به ClpP میتوکندری میچسبد و در نتیجه کنترل کیفیت پروتئینهای میتوکندریایی دچار اختلال میشود. میتوان ClpP را مانند سیستم دفع زباله در نیروگاه تصور کرد؛ زمانی که این سیستم بسته میشود یا از کار میافتد، پسماند تجمع مییابد و موتور دچار اختلال میشود. پیامدهای بعدی برای هر کسی که بیماری پارکینسون را مطالعه کرده آشناست: نورونها کارایی خود را از دست میدهند، مدارهای تولید دوپامین کمکار میشوند، التهاب افزایش مییابد و حرکت و تواناییهای شناختی شروع به افت میکنند.
بهجای حمله گسترده به آلفا-سینوکلئین، محققان یک پپتید کوتاه به نام CS2 طراحی کردند تا بهعنوان طعمه عمل کند. ایده ساده و در عین حال زیباست: پروتئین مهاجم را بهسمت طعمه بکشانند تا از نزدیکی ClpP دور شود و میتوکندریها بتوانند وظایف خود را انجام دهند. در بافتهای مغزی انسانی خارجشده (ex vivo)، در نورونهای کشتشده، و در مدلهای موشی، CS2 نشان داد که نشانگرهای التهاب را کاهش میدهد، فعالیت میتوکندری را حفظ میکند و بهبودهای قابلاندازهگیری در آزمونهای حرکتی و شناختی حیوانات بهوجود آورد.
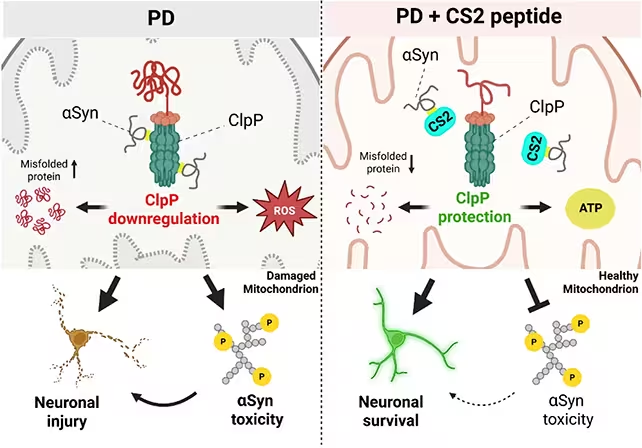
نشان داده شد که CS2 از آنزیم ClpP محافظت میکند.
این اثر محافظتی اهمیت زیادی دارد زیرا به یک گام مکانیکی هدف میزند که تصور میشود در رأس بسیاری از ناکامیهای سلولیِ مرتبط با پارکینسون قرار دارد. اگر تعامل آلفا-سینوکلئین با ClpP یکی از نخستین اختلالات در آبشار بیماری باشد، محافظت از ClpP میتواند از مجموعهای از آسیبهای بعدی جلوگیری کند یا دستکم سرعت آنها را کاهش دهد.
برای تبیین بیشتر یافتهها، تیم تحقیقاتی از ترکیبی از تکنیکهای زیستشناسی مولکولی استفاده کرد؛ از آن جمله ردیابی تعاملات پروتئین-پروتئین با روشهای ایمونوافلورسانس و کوایمیون، آنالیزهای فعالیت پروتئازی ClpP، اندازهگیریهای مصرف اکسیژن سلولی (OCR) برای ارزیابی تنفسی میتوکندری و آزمونهای رفتاری استاندارد در موشها. این ترکیب روشها به محققان امکان داد تا از سطح مولکولی تا سطح عملکردیِ نورونها را دنبال کنند و همبستگی مستقیمی میان پیوند آلفا-سینوکلئین به ClpP و نارسایی متابولیک نورونها برقرار کنند؛ بدین ترتیب فرضیه علت-معلولی تقویت شد.
علاوه بر این، در آزمایشهای کشت سلولی تیم نشان داد که حضور CS2 مانع اتصال آلفا-سینوکلئین به ClpP میشود و در نتیجه نمایهای از تحمل سلولی بهتر و پارامترهای متابولیک پایدارتر پدید میآید. این نتایج، بهویژه در نورونهای دوپامینیساز، اهمیت بالایی دارد زیرا این جمعیت سلولی بیشترین آسیب را در بیماری پارکینسون متحمل میشوند.
زمینه علمی و پیامدها
پارکینسون یک اختلال تکمسیره نیست؛ ژنها، محیط، پیری و فشارهای سلولی با هم در تعاملاند و نتیجه مجموعهای از مسیرهای مولکولی پیچیده است. این مطالعه مدعی درمان قطعی جهانی نیست، بلکه وضوحی عرضه میکند: یک نقطه تماس مولکولی مشخص که میتوان آن را تغییر داد. مداخله در این نقطه میتواند مکمل رویکردهایی باشد که تجمع آلفا-سینوکلئین را کاهش میدهند، تولید میتوکندری جدید (بیوژنز میتوکندری) را تقویت میکنند یا التهاب را تنظیم میکنند.
تیم پژوهشی برآورد میکند که حدود پنج سال زمان لازم است تا CS2 یا زیستداروهای مشابه ممکن است به اولین آزمایشهای انسانی برسند. این زمانبندی برای یک درمان مبتنی بر پپتید منطقی است، اما احتیاط لازم است. هر مولکولی که بر تعاملات پروتئینی درون میتوکندری تأثیر میگذارد باید بهطور جامع از نظر اثرات خارجهدف (off-target) و پیامدهای احتمالی ایمنی بررسی شود. تغییرات کوچک در کنترل کیفیت پروتئینها میتواند به نتایج غیرمنتظرهای منجر شود، مانند تغییرات ناخواسته در پروتئینهای دیگر یا تداخل با مکانیسمهای بازسازی میتوکندری.
با این حال، این مطالعه بهطور نسبی صفحات تکتونیکی پژوهش پارکینسون را کمی جابهجا میکند. به دانشمندان مکانیزمی قابل هدف و یک مولکول نمونهاولیه ملموس برای پالایش میدهد. برای خانوادههایی که با این بیماری زندگی میکنند، آن وضوح جزئی معنادار است؛ تداوم درمانهایی که متابولیسم انرژی نورونها را حفظ میکنند میتواند به زندگی طولانیتر و با کیفیت عملکردی بهتر منجر شود.
در زمینه پژوهشهای بالینی و ترجمهای، نکاتی عملی وجود دارد که باید در نظر گرفته شوند: پایداری پپتید CS2 در محیط بدن، نفوذپذیری خون-مغز (BBB) و انتقال ایمن و مؤثر آن به نورونهای هدف، زمانبندی درمان (پیش از بروز علائم یا پس از تشخیص) و امکان ترکیب با داروهای حمایتی مانند آگونیستهای دوپامین یا درمانهای ضدالتهابی. همچنین نیاز به مدلسازی فارماکوکینتیک/فارماکودینامیک (PK/PD) دقیق و ارزیابی دوز-پاسخ در مدلهای پیشبالینی وجود دارد.
از منظر زیستمولکولی، یافتهها سوالات جدیدی نیز مطرح میکنند: کدام گونههای آلفا-سینوکلئین (مونومرها، الیگومرها یا فیبریلها) بیشترین گیرش به ClpP را دارند؟ آیا تغییرات پساتراُش (post-translational modifications) در آلفا-سینوکلئین یا در خود ClpP بر این تعامل تأثیر میگذارد؟ و آیا جهشهای ژنتیکی مرتبط با پارکینسون مستعدسازی این تعامل مخرب را افزایش میدهند؟ پاسخدادن به این پرسشها میتواند راه را برای درمانهای هدفمندتر باز کند.
دیدگاههای تخصصی
«این کار ترکیبی نادر از دقت مکانیکی و خلاقیت درمانی است»، میگوید دکتر النا رامیرز، یک نوروبیولوژیست (نمونهای صریح)، که روی دینامیک میتوکندری مطالعه میکند. «طراحی یک طعمه برای رهگیری یک پروتئین بیماریزا در آستانه میتوکندری هوشمندانه است. چالش پیشروی ما رساندن مقدار کافی از پپتید به نورونهای آسیبپذیر بدون ضعیفکردن سیستمهای کنترل کیفیت سلولی دیگر است.»
این مقاله در نشریه Molecular Neurodegeneration منتشر شده و چندین مسیر پژوهشی جدید را باز میکند: بهینهسازی پایداری CS2، آزمونهای ایمنی طولانیمدت، و بررسی اینکه آیا طعمههای مشابه میتوانند از آنزیمهای میتوکندریایی دیگر نیز محافظت کنند. پرسش بزرگتر این است: اگر بتوانیم از خالیشدن انرژی میتوکندری جلوگیری کنیم، آیا میتوانیم مسیر پارکینسون را از یک فرایند پیشرونده بیوقفه به حالتی قابل مدیریت یا حتی متوقفشدنی تغییر دهیم؟ پاسخ آزمایشهای آینده تعیینکننده خواهد بود، اما در حال حاضر میدان پژوهش یک سرنخ تازه برای پیگیری در اختیار دارد.
در نهایت، یافتهها بر اهمیت تمرکز بر متابولیسم سلولی و کیفیت پروتئینهای میتوکندریایی بهعنوان اهداف درمانی تأکید میکنند. ترکیب رویکردهای مولکولی مانند طعمههای پپتیدی با استراتژیهای کلیتر شامل اصلاح سبک زندگی، درمانهای دارویی موجود و تکنیکهای پیشرفته مانند درمانهای ژنی میتواند نقشه راهی چندگانه برای مقابله با پارکینسون فراهم آورد.
برای ارتقای این خط تحقیق به مرحله کاربردی، همکاری میان تیمهای بیوانفورماتیک، زیستشناسی مولکولی، داروسازی و بالین ضروری است. تحلیلهای ساختاری دقیقتر میتواند محل اتصال آلفا-سینوکلئین به ClpP را با وضوح اتمی مشخص کند و امکان طراحی پپتیدهای جدید با نفوذپذیری بهتر، پایداری طولانیتر و عوارض جانبی کمتر را فراهم آورد. همچنین مطالعات همکار با بانکهای بافت انسانی و مجموعههای بالینی پارکینسون میتواند اعتبار بالینی این هدف را تقویت کند.
در مجموع، این تحقیق نه تنها یک هدف مولکولی مشخص ارائه میدهد، بلکه یک مسیر منطقی برای ترجمه علمی به درمان بالقوه را نیز نمایان میسازد؛ موضوعی که میتواند در سالهای آتی تأثیر مهمی بر پژوهشها و توسعه درمانهای نوین برای بیماری پارکینسون داشته باشد.








.webp "نخستین نشانه جدی از قمر فراخورشیدی عظیم دوردست")



نظر بگذارید
نظرات (5)
اگر CS2 بشه پایدار و ایمن، میتونه تحول باشه. البته ۵ سال؟ کاش زودتر، منتظر خبرهای بالینیتریم 🙂
ایده خلاقانه اما واقعیت اینه: پپتیدها تو بدن ناپایدارن، نفوذ به مغز سخت، امیدوارم تیم راه حل داشته باشه
این واقعا تو همه مراحل پارکینسون نقش داره یا فقط تو مدل حیوانی؟ دادهی انسانی کسی دیدی؟
منطقیش اینه که هدف گیری ClpP میتونه مکمل درمانهای دیگه باشه، ولی عوارض و خارجهدفیش رو واقعا میترسم...
وااای، اینکه آلفا-سینوکلئین مستقیم به میتوکندری میزنه شوکهکننده بود! امیدبخش اما باید ببینیم تو انسان چطوره، دعا میکنم